Article Text

Abstract
Rationale Bronchiectasis is characterised by excessive production of mucus and pulmonary exacerbations. Inhaled osmotic agents may enhance mucociliary clearance, but few long-term clinical trials have been conducted.
Objectives To determine the impact of inhaled mannitol on exacerbation rates in patients with non-cystic fibrosis (CF) bronchiectasis. Secondary endpoints included time to first exacerbation, duration of exacerbations, antibiotic use for exacerbations and quality of life (QOL) (St George's Respiratory Questionnaire, SGRQ).
Methods Patients with non-CF bronchiectasis and a history of chronic excess production of sputum and ≥2 pulmonary exacerbations in the previous 12 months were randomised (1:1) to 52 weeks treatment with inhaled mannitol 400 mg or low-dose mannitol control twice a day. Patients were 18–85 years of age, baseline FEV1 ≥40% and ≤85% predicted and a baseline SGRQ score ≥30.
Main results 461 patients (233 in the mannitol and 228 in the control arm) were treated. Baseline demographics were similar in the two arms. The exacerbation rate was not significantly reduced on mannitol (rate ratio 0.92, p=0.31). However, time to first exacerbation was increased on mannitol (HR 0.78, p=0.022). SGRQ score was improved on mannitol compared with low-dose mannitol control (−2.4 units, p=0.046). Adverse events were similar between groups.
Conclusions Mannitol 400 mg inhaled twice daily for 12 months in patients with clinically significant bronchiectasis did not significantly reduce exacerbation rates. There were statistically significant improvements in time to first exacerbation and QOL. Mannitol therapy was safe and well tolerated.
Trial registration number NCT00669331.
- Bronchiectasis
- Respiratory Infection
Statistics from Altmetric.com
Introduction
Bronchiectasis is a chronic lung condition in which damage to the airways causes abnormal dilatation of the bronchi and impaired mucociliary clearance.1 The increase in mucus accumulation is accompanied by chronic cough and recurrent infections, often resulting in significant morbidity and mortality.1 ,2 The incidence of non-cystic fibrosis (CF) bronchiectasis varies from approximately 3.7/100 000 to 1470/100 000 per year in remote and less affluent populations.3–5 However, due to its coexistence with other chronic respiratory diseases, many people with non-CF bronchiectasis remain undiagnosed and undertreated.1 ,6
Airway clearance techniques are recommended.7 ,8 Pharmacological agents, such as mucolytics, osmotic agents that increase airway hydration, bronchodilators and antibiotics may be employed, but few well-controlled clinical studies have investigated these agents.7 ,9–15
Mannitol is a naturally occurring sugar alcohol.9 When inhaled it improves mucus clearance both acutely and over 24 h in non-CF bronchiectasis and improves mucus clearance and FEV1 in patients with CF.10 ,11 ,16–19 It is likely that water drawn into the airway lumen by osmotic gradient favourably changes mucus properties.9 ,17 ,20 Improvement in hydration, reduction in surface tension and rheological properties of mucus, are postulated to increase mucociliary and cough clearance.
Two open-label pilot studies reported that mannitol at a dose of 400 mg reduced the surface tension of mucus, improved the function of small airways and improved the health-related quality of life in a small group with bronchiectasis.9 ,12 ,21 A placebo-controlled study showed a significant change in sputum weight over a period of 12 weeks, with associated lower antibiotic use. In a subgroup with high-resolution CT (HRCT) scanning, the mannitol users showed a reduction in small airway mucus plugging.22
The aim of this randomised, controlled, double-blind study was to determine the impact of inhaled mannitol on exacerbation rates and quality of life in patients with non-CF bronchiectasis.
Methods
This was a 52-week, double-blind, randomised, controlled trial of inhaled mannitol (Pharmaxis, Frenchs Forest, New South Wales, Australia), 400 mg twice a day, versus a control of inhaled mannitol, 50 mg twice a day. Respirable spray-dried mannitol of 3 µ diameter was used in both arms. Low-dose respirable mannitol was chosen as the control since it maintains the same taste and sensation characteristics and since a previous dose-ranging study in CF-related bronchiectasis found no clinical benefit of mannitol at this dose.23
Selection of patients
The study was conducted in full accordance with the current revision of the Declaration of Helsinki and the Good Clinical Practice: Consolidated Guideline approved by International Conference on Harmonisation (ICH). The study was approved by the institutional review board or ethical committee of each participating hospital, and written consent was obtained from each patient or their legally authorised representative.
The study was conducted at 84 sites in the USA (20), Europe (34), Australia (10), New Zealand (3) and South America (17). To be eligible, patients had to be aged 18–85 years, with a HRCT confirmed diagnosis of non-CF bronchiectasis, baseline FEV1≥40% and ≤85% predicted and ≥1 L and a baseline St George's Respiratory Questionnaire (SGRQ) score ≥30. Patients had to have had a minimum of two pulmonary exacerbations in the previous 12 months and at least four exacerbations in the previous 2 years. Pragmatically, any cause of bronchiectasis (other than CF) was deemed eligible. Individuals were given a tolerance test of 400 mg inhaled mannitol (MTT) to exclude those with mannitol-induced bronchospasm. The use of low-dose mannitol in the control arm allowed masking of treatment arm to be continued after MTT. Use of nebulised hypertonic saline was prohibited to avoid confounding, but all other therapies were continued. (Full details of the inclusion–exclusion criteria and the MTT can be found in the online supplement ‘Study population and MTT’).
Randomisation and masking
This was a double-blinded study. Patients were randomised to active treatment or the control arm in a 1:1 ratio. Randomisation was stratified by site, in a permuted-block design.
Study drug
Patients were assigned to receive 10 capsules of inhaled mannitol, either 40 mg (mannitol group) or 5 mg per capsule (control group) twice a day for 52 weeks. The drug was administered by a single-dose dry-powder RS01 inhaler Model 7 (Plastiape, Milan, Italy). The patients had two screening visits, six visits over the 52-week on-treatment period and a further 4-week follow-up visit (figure 1).

Study design.
Outcome measures
The primary efficacy endpoint of the study was pulmonary exacerbation rate, defined as the number of all pulmonary exacerbation events observed for a patient within one treatment year. A pulmonary exacerbation was pragmatically defined as a worsening in signs and symptoms requiring a change in treatment (derived from Anthonisen).24 Additionally, exacerbations were graded for severity as a sensitivity measure (see online supplement ‘Classification of Graded Pulmonary Exacerbations’). Treatment of exacerbations including type and duration of antibiotic was left to the discretion of the investigator.
Secondary endpoints included comparisons between treatment groups for time to first exacerbation, duration of exacerbations, quality of life using the SGRQ, antibiotic use for exacerbations, 24 h sputum weight, change in FEV1 and FVC. Number of exacerbation-related hospitalisations was a preplanned exploratory analysis. To ensure consistency, each patient was provided an Acapella mucus mobilisation device.
Safety
Safety was assessed by monitoring of adverse events (AE), complete blood count, liver and renal function tests, qualitative sputum microbiology and physical examinations including vital signs.
Statistics and statistical methods
The study was designed to have 80% power to detect a 27% reduction in pulmonary exacerbation event rate, leading to a sample size of 237 per group. This provided 80% power to detect a two-unit difference in SGRQ score (see online supplement ‘Sample Size and Power Calculations’).
The primary efficacy endpoint was graded pulmonary exacerbation rates. The rate was defined as the number of all graded pulmonary exacerbation events observed within one treatment year. A negative binomial model including treatment, geographical region and baseline exacerbation rate as predictors, with the log of follow-up time as the offset, was used to analyse pulmonary exacerbation rate, number of antibiotic-treated exacerbations, the number of days on antibiotics for the treatment of exacerbations, number of days with exacerbations and number of hospitalisations due to exacerbations. SGRQ, 24 h sputum weight and lung function were analysed using general linear mixed model repeated measures methodology and an unstructured variance–covariance structure. Time to first exacerbation was analysed using the Cox proportional hazard method.
Results
The study was conducted between October 2009 and February 2013. Patient disposition is shown in figure 2. Four hundred and eighty-six (84%) persons passed the MTT, 485 were randomised and 461 received blinded study treatment. Patient demographics were balanced at baseline table 1. Overall, study patients had a mean baseline rate of pulmonary exacerbations of 3.22 events/year (3.20 events/year mannitol group; 3.25 events/year control group).
Baseline characteristics of intention-to-treat population

Subject disposition. MTT, mannitol tolerance test.
Compliance and completion
Compliance was calculated from the proportion of used allocated capsules. Two hundred and twenty patients in the mannitol arm and 215 patients in the control arm had compliance data and mean compliance was 92.7% (SD 13.4%) and 94.2% (SD 9.2%), respectively.
All visits were completed by 191 (82.0%) patients in the mannitol arm and 189 (82.9%) in the control. Not all continued with study drug throughout; 74% patients in the mannitol and 76% in the control arm completed at least 50 weeks of study treatment.
Sixteen (6.9%) patients in the mannitol arm and 10 (4.4%) in the control withdrew due to AEs.
There were slightly more withdrawals in the first 3 months in the mannitol arm (7.3%) than in the low-dose mannitol control arm (4.4%). Later withdrawal rates were balanced between arms.
Efficacy
Primary endpoint
The annual rate of exacerbations in the mannitol and control arms were 1.69 (95% CI 1.48 to 1.94) and 1.84 (95% CI 1.61 to 2.10), respectively. The rate ratio calculated from the negative binomial regression model was 0.92 (95% CI 0.78 to 1.08) and was not statistically significant (p=0.31).
Secondary endpoints
Secondary endpoint results are summarised in table 2.
Summary of results for key secondary efficacy endpoints
Time to first exacerbation
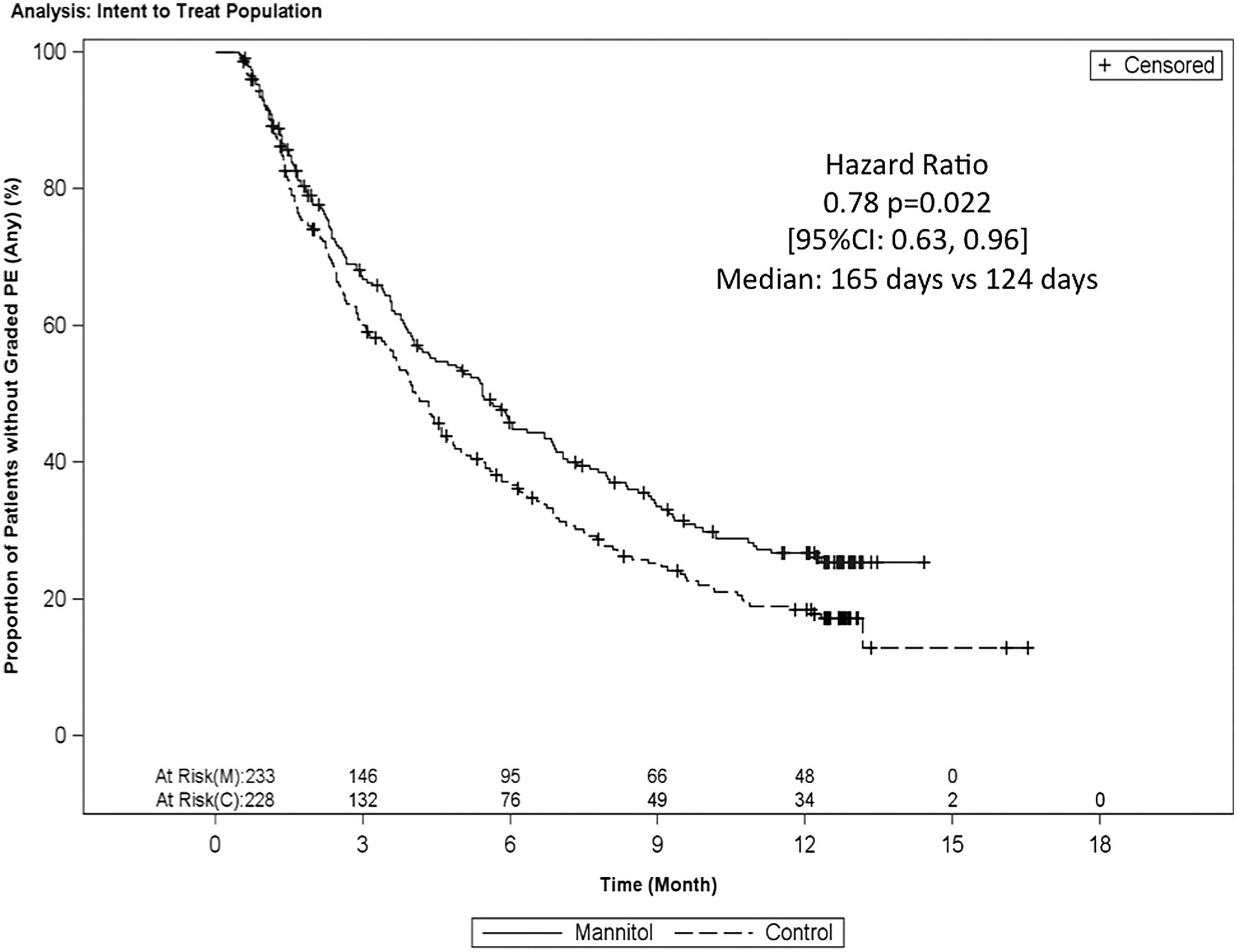
Time to first exacerbation was longer in the mannitol versus control arms (165 days vs 124 days, p=0.021 (log-rank) (figure 3). The Cox regression-derived HR was 0.78 (0.63 to 0.96) which was statistically significant (p=0.022). 31.3% of mannitol users and 21.9% of control patients remained exacerbation free throughout the 12-month study (posthoc, risk ratio 0.88, p=0.027).

Kaplan–Meier plot of the time to first graded pulmonary exacerbation.
Antibiotic use and hospitalisations for exacerbations
The number of days of antibiotic used to treat exacerbations was reduced by 24% (p=0.0496) from a mean 26.03 (95% CI 21.11 to 32.09) days per year in the control arm to a mean 19.88 (95% CI 16.12 to 24.51) days in the mannitol arm. Although hospitalisation rate due to pulmonary exacerbation was 31% lower in the mannitol arm (0.14 hosp/year) than the control (0.20 hosp/year), this difference was not significant (p=0.1798).
St George's Respiratory Questionnaire
The change in total SGRQ score over the treatment period was −10.98 (95% CI −12.78 to −9.18) and −8.58 (95% CI −10.43 to −6.72) for mannitol and control, respectively. The difference of −2.40 units between arms was statistically significant (p=0.046). The changes in the individual domains (Activity, Impact and Symptoms) also all trended in favour of mannitol (figure 4). The Activity score was significantly greater on mannitol (p=0.0339).

St George's Respiratory Questionnaire (SGRQ) domain scores.
24 h sputum weight
Mean 24 h sputum weight fell progressively over the course of the study in both treatment arms, but remained higher in the mannitol arm than the control arm throughout. In the mannitol arm, there was a least-squares mean reduction in sputum weight for the on-treatment period of 6.6 g, compared with a reduction in sputum weight of 9.42 g in the control arm (difference=2.76 g; p=0.035).
Spirometry
There were no significant changes in lung function in either arm. The least-square mean changes in FEV1 and FVC over the treatment period for mannitol and control were 2.36 (95% CI −24.03 to 28.74) mL, 0.15 (95% CI −36.75 to 37.05) mL and −5.20 (95% CI −32.35 to 21.95) mL, −15.70 (95% CI −53.64 to 22.25) mL, respectively.
Safety
MTT findings
Patients undertook the MTT without prior bronchodilator use; 96 of 581 (16.5%) patients failed the test (>20% fall in FEV1 or unable to complete test) and, therefore, were not eligible. AEs occurring on the day of or day after the MTT were reported by 21.7% of patients. Events possibly causally related to the MTT were reported in 7.2%; the most common were cough (3.1%) and fall in FEV1 (2.2%). Bronchospasm (0.5%), oxygen desaturation (0.7%) and self-reported wheeze (0.5%) were uncommon.
Adverse events on treatment
The 12 most frequently reported AEs are listed in table 3. The majority of AEs were judged mild or moderate; only 21.5% patients in the mannitol arm and 28.1% patients on control experiencing severe AEs. AEs leading to study withdrawal were slightly more frequent in the mannitol (6.9%) than the control arm (5.3%); 20.2% patients in the mannitol arm and 16.7% on control had AEs considered causally related to study medication. Serious adverse events (SAE) were less frequent in the mannitol arm (18.5% vs 22.4%); most frequent were ‘condition aggravated’ due to exacerbation (mannitol 9.0%, control 11.4%). Most SAEs were reported by <1% of patients. Two deaths occurred, both on control.
12 most commonly reported adverse events by treatment group
Blood and sputum analysis
No significant pattern of change was detected in either haematology or biochemistry values (data not shown). At baseline, 52.7% patients in the mannitol arm and 50.5% patients in the control, had potentially pathogenic organisms in their sputum. At week 52, the proportion of patients with abnormal flora had fallen by 13.1% versus 8.3% in the mannitol and control arms, respectively. There was no increase in infection with any specific pathogen.
Discussion
We have reported the largest randomised controlled trial of a mucoactive therapy in non-CF bronchiectasis to date. We found that the annual exacerbation rate was reduced in both the high dose (400 mg twice daily) and low dose (50 mg twice daily) control arms during the course of the study, although there was no difference in annual exacerbation rate between the two arms. However, inhaled mannitol at a dose of 400 mg twice daily significantly extended the time to first exacerbation, increased the proportion of patients who remained exacerbation free, reduced the number of days of antibiotic therapy and improved quality of life.
Although the study cohort was intentionally enriched for a subset of adults with bronchiectasis having a significant impact on their lives, including having at least two exacerbations in the previous 12 months, a minimum SGRQ score of 30, producing at least 10 grams of sputum a day and an FEV1 <85% predicted, the population studied is arguably representative of many non-CF bronchiectasis patients seen in secondary care. Patients experienced, on average, more than three exacerbations in the previous year (similar to rates in a recent national UK audit), and approximately 20% of patients were colonised with Pseudomonas aeruginosa infection, which was slightly lower than expected.25 ,26
Despite the favourable effects on a number of clinical parameters, the primary endpoint was not met with the rates of exacerbations in both arms being less than their reported historical rates.27 This lack of effect did not appear to be caused by high event rates in a subset of mannitol users (data not shown). The control was inhaled mannitol in a dose of 50 mg, which, in a previous dose-ranging study in CF, did not show any effect on FEV1 after 2 weeks of treatment.23 Whether lack of efficacy of low-dose mannitol applies in non-CF bronchiectasis over 12 months is unknown, a control effect cannot be excluded. Alternately, since the study was powered to detect a treatment effect of 27%, assuming the annualised exacerbation rate remained similar to historical rates, it is possible that the study suffered from a type 2 error. Finally, it is acknowledged that exacerbation rate is a relatively blunt tool when the aim is to discern differences in relatively infrequent events. The use of time to first event is being increasingly recognised (including by some regulatory authorities), being less reliant than change in exacerbation rate on subjective judgement to estimate the end of the event.
The SGRQ is validated in bronchiectasis.28 The difference between the arms was significant (estimated difference 2.5, p=0.05), although the CI was wide. The Activity domain was in itself significantly improved, which reflects an improvement in limitations due to breathlessness or ability to carry out more activities that caused breathlessness.
The time to first exacerbation was significantly delayed by 22% with the longer time to first event being driven by both a lower incidence of exacerbations and an increased time to first event in those having an exacerbation in the mannitol arm. Patients in the mannitol arm also had significantly fewer days (24%, p=0.0496) on antibiotics for the treatment of exacerbations, as a result of both shorter and fewer antibiotic-treated exacerbations. However, the CI for the treatment difference was wide and only just excluded zero. There was also a longer median time to first antibiotic use for all treated pulmonary exacerbations (158 days vs 122 days, p=0.021), confirmed by Cox regression (HR 0.78, p=0.02). The study was not powered to properly evaluate hospitalisations. Nevertheless, the positive trend favouring mannitol was also consistent with the overall exacerbation-related data.
In this study, the end of an exacerbation could be immediately followed by another one. The major impact of varying the definition of the end of an exacerbation on the overall exacerbation rate is demonstrated by a posthoc exploration of separating events by an arbitrary 2-week period between the end of one and the start of another exacerbation, which has commonly been applied in other respiratory studies,29 ,30 with 21.5% of exacerbations in the mannitol arm and 14.1% on control which occurred within 15 days of each other. After forcing distinct separation of events, the difference in rate grew (rate ratio 0.88, p=0.086). This may, in part, explain the anomaly of meaningful improvements in antibiotic use, time to first exacerbation and exacerbation incidence despite an apparent lack of effect on rate.
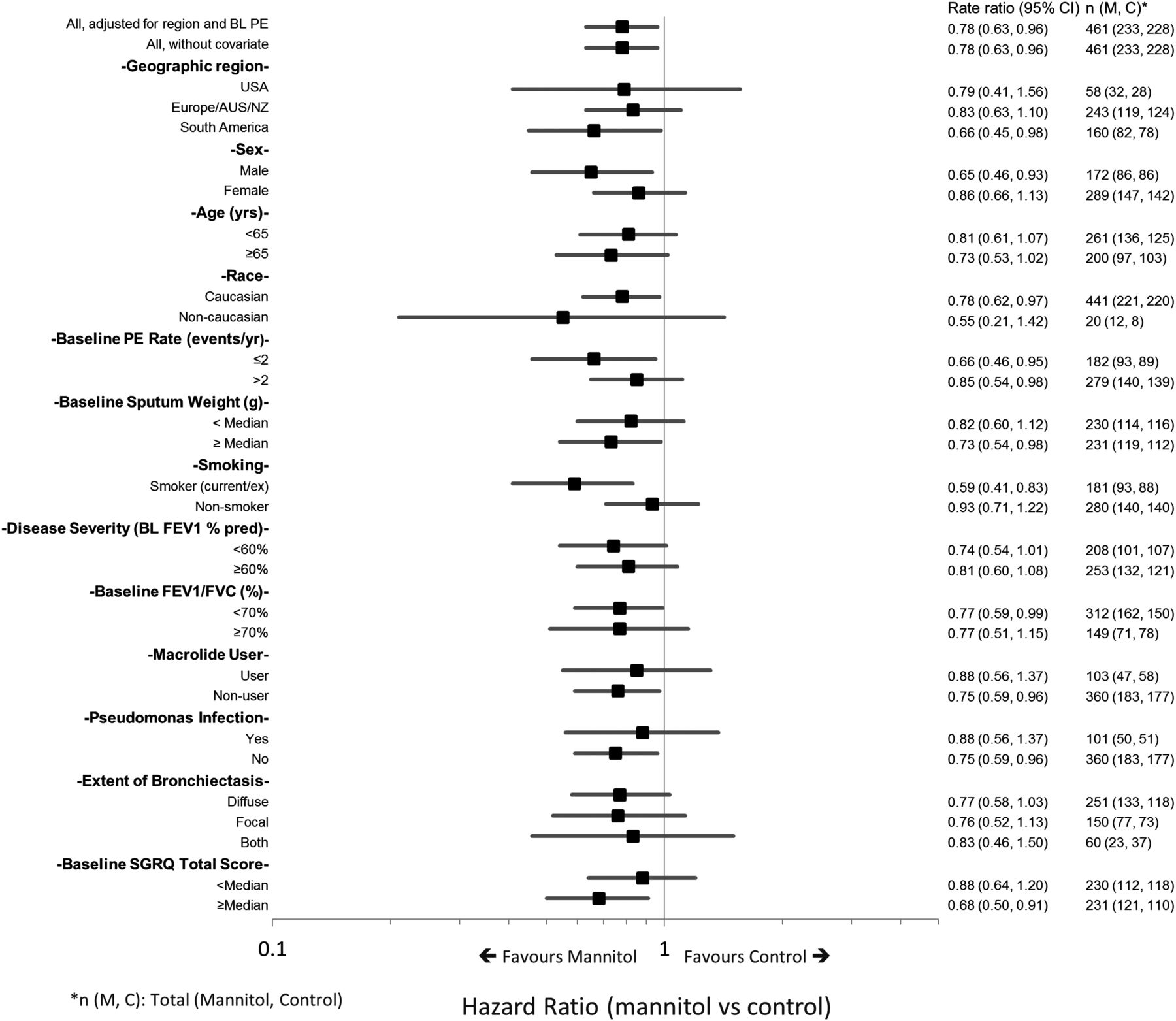
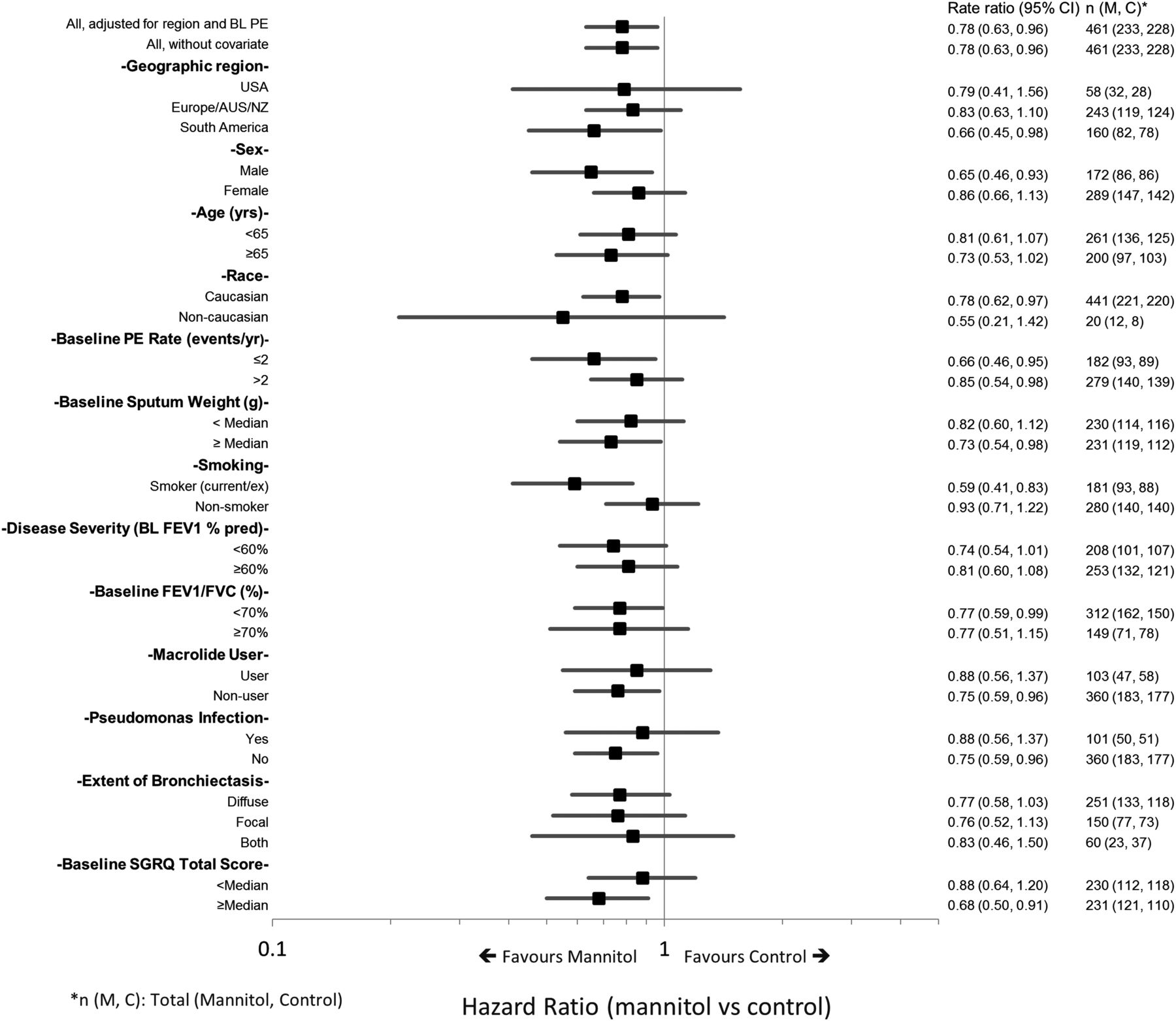
A preplanned analysis of time to first exacerbation in subgroups was undertaken (figure 5). This suggested an increased benefit for mannitol over control for patients who had poorer quality of life at baseline (SGRQ greater than or equal to the median) (HR=0.68; 95% CI 0.50 to 0.91), two pulmonary exacerbations per year at baseline (HR=0.66; 95% CI 0.46 to 0.95), or who were current/ex-smokers (HR=0.59; 95% CI 0.41 to 0.83).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Forest plot of HR for time to first exacerbation by predefined subgroup.
Mannitol increases mucociliary clearance and, therefore, would be expected to lead to a greater 24 h sputum weight.9 ,19 ,31 The decreases from baseline in sputum weight were difficult to interpret. The pattern of declining sputum weight seen over time in both groups may reflect improved clearance through study involvement and routine use of Acapella, but could suggest an effect of low-dose mannitol.
We did not see any difference in FEV1. This finding is in contrast with CF where mannitol is known to improve FEV1.32 This observation, together with the other results of this trial, highlight that non-CF bronchiectasis represents a distinct clinical entity, so that an evidence base for therapy cannot simply be adopted from that developed for CF nor should trial design be similarly transposed.
In conclusion, this study of mannitol inhaled twice daily for 12 months in patients with mild to moderate bronchiectasis did not lead to a significant reduction in the exacerbation rate. Despite this, there were significant improvements in time to first exacerbation, duration of antibiotic use and quality of life. Mannitol therapy was safe and well tolerated over a period of 12 months.
Acknowledgments
The authors are indebted to the investigators, centre personnel and study participants for their active contributions to the study and to Zoe Silverstone for writing expertise. Infrastructure supporting recruitment in the UK was provided by the National Institute for Health Research Comprehensive Local Research networks (NIHR-CLRN). Dianna Bilton is supported by the NIHR Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London. Tobias Welte is a member of the German Center of Lung Research.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online supplement
- Data supplement 3 - Online supplement
Footnotes
Contributors All authors helped to interpret data, participated in drafting/writing/review of the manuscript, and have seen and approved the final version. DB was Principal Investigator for the study, participated on the Steering Committee for the design of the study, and had full access to all the data in the study, and final responsibility to submit for publication. GT and AFB were on the Steering Committee for the design of the study. GT, ADS, DCC, LJAD, CO'D, EHJvH, LOV, TW were all investigators and or country Leads for the study. BC was the Sponsor's Responsible Medical Officer, designed the study with the Steering Committee, approved the protocol and SAP and assisted with statistical plans and analysis. HGF assisted in the study design, statistical plans and analysis. JW wrote the Statistical Analysis Plan and was responsible for the data analysis for the study.
Funding The study was funded by Pharmaxis, Frenchs Forest, NSW Australia.
Competing interests None.
Patient consent Obtained.
Ethics approval Ethics approval was obtained from Research Ethics Committees for all participating centres.
Provenance and peer review Not commissioned; externally peer reviewed.